Computer Code Solves a Quantum Mechanical Problem
Focusing on the strange and surprising quantum world, a unique computer code that can model systems comprising thousands of atoms is driving fresh discoveries in areas as diverse as new materials, clean energy, nanotechnology and pharmaceuticals.
Called ONETEP (Order-N Electronic Total Energy Package), the code plugs the critical gap between what can be achieved by state-of-the-art experiments and by conventional quantum-level computer modelling & simulation of just a few hundred atoms at a time.
This is a classic story of how multi-institution, multidisciplinary co-operation – in this case including SES members Cambridge and Southampton Universities and Imperial College London – can build, extend and exploit a game-changing, world-leading research tool.
A Major Step for Modelling
Quantum mechanics – the theory governing matter’s structure and behaviour at the very smallest scale – is a branch of science vital to the relentless quest for progress. In this, the realm of atomic and subatomic particles, computational tools play a key role in exploration and investigation, with modelling & simulation producing insights of huge value in fields such as drug design and the development of ‘metamaterials’ with properties not found in nature.
For around 30 years, DFT (Density Functional Theory) has been one of the most widely used materials modelling methods at the atomic scale. Crucially, however:
- Computational costs ramp up substantially every time an atom is added to a ‘virtual’ sample being examined – for instance, making the system studied 10 times bigger increases the computational cost 1000-fold.
- This means conventional DFT simulations have been restricted to a few hundred atoms, but much larger simulations are needed in order to produce reliable results that can open doors to advances with genuine real-world benefits.
ONETEP squarely addresses this limitation, enabling DFT simulations of up to 100,000 atoms but without any explosion in computational costs. As well as revealing electronic structures, the data produced by ONETEP can be processed to generate a range of information about how matter behaves and bonds at the quantum scale, enabling investigation of an extraordinary array of phenomena, processes, molecules and materials.
The first steps in the ONETEP story were taken in the 1990s by the Theory of Condensed Matter (TCM) Group at Cambridge’s Cavendish Laboratory. As those involved moved on to independent academic positions, so the number of institutions contributing to ONETEP’s development expanded to include Southampton and Imperial, with Warwick University more recently coming on board. Along the way, long-term support from the Engineering and Physical Sciences Research Council (EPSRC) has underpinned the project through software development grants and a Platform Grant.
Professor Peter Haynes, now at Imperial, was one of the code’s original authors and remains a key player in the ONETEP Developers’ Group.
“Developing ONETEP has meant building a balanced team not just across different disciplines but also across different institutions“
Professor Peter Haynes
“Developing ONETEP and continuously extending its capabilities and applications has only been possible by pooling many skillsets, strengths and specialisms,” he says. “It’s meant building a balanced team not just across different disciplines such as physics, chemistry, biochemistry, materials science, engineering and computer science, but also across different institutions.”
From Quantum World Insights to Real World Impacts
Now firmly established as a hugely effective modelling and simulation tool, ONETEP is marketed as part of the Materials Studio suite of software with a graphical interface and full support from Dassault Systèmes BIOVIA; inexpensive licenses are available for academic use across the world. To maximise its value and versatility, substantial effort has been invested in optimising the code for supercomputing environments and enabling it to function with its trademark accuracy and reliability on massively parallel processing architectures.
Some past examples of the variety of projects with ONETEP include:
Dr Tim Zuehlsdorff – TCM Group, Cambridge
“I worked on ONETEP as a PhD student at Imperial. Later at Cambridge, I used the code to study the large protein complexes often found in bacteria that harvest light to meet their energy needs. This work was enhancing our understanding of photosynthesis to aid the development of next-generation solar cells – including more efficient devices inspired by nature.”
Dr Fabiano Corsetti – Department of Materials, Imperial
“As part of my PhD, I wrote a new ONETEP module for calculating materials’ vibrational frequencies. I then went on to using ONETEP to study the electronic properties of graphene when the surface of this ‘miracle material’ had been doped with different atoms and molecules. ONETEP delivers a detailed, atomic-level interpretation of what’s happening in our physical experiments.”
Dr James Womack – School of Chemistry, Southampton
“The description of complicated interelectronic interactions is a crucial component of accurate simulations of matter. ONETEP offers many sophisticated models for the interactions of electrons and I worked on extending these capabilities by implementing highly accurate models which make use of a quantity known as the ‘kinetic energy density’. These new capabilities add to the already significant theoretical toolbox available to scientists using ONETEP in their research.”
Dr Niccolò Corsini – Department of Materials, Imperial
“Having worked on ONETEP during my PhD, I later used it to model the optical properties of nanocrystals. It’s been particularly good at disentangling the effects of pressure, size and ligands (ions and molecules that perform bonding functions). The findings have potential to help develop innovative pressure sensors and ‘tunable’ solar photovoltaic devices.”
Dr José María Escartín Esteban – TCM Group, Cambridge
“My aim was to incorporate spin-orbit couplings into ONETEP so the code can, for example, better describe materials where atoms with high atomic numbers are present. This will enable the study of next-generation low-power electronic devices. The ONETEP team displays a superb level of co-ordination and collaboration in software development and resource sharing.”
Partner Power
The ONETEP story is an excellent example of the value of partnership – and of how close co-ordination and integration of skills and disciplines can make a real difference to devising a powerful solution to a pressing research need.
“Annual coding ‘retreats’ enable all of us on the ONETEP team to go on sharing ideas, expertise and experiences as part of the process of continuously improving the code,” says Dr Jacek Dziedzic of Southampton’s School of Chemistry. “The aim is to keep adding more applications and functionality to ONETEP, in line with the expectations of DFT users. The ultimate goal is to ensure that what ONETEP enables is limited only by the availability of computational power and by the imagination of researchers themselves.”
Current members: Dr Nicholas Hine (Warwick), Dr Jacek Dziedzic (Southampton), Professor Arash Mostofi (Imperial), Professor Chris-Kriton Skylaris (Southampton), Professor Peter Haynes (Imperial) and Professor Mike Payne (Cambridge).
Further Information
- ONETEP website: onetep.org
- ONETEP datasheet: http://accelrys.com/products/datasheets/onetep.pdf
- TCM Group, Cavendish Laboratory, Cambridge: tcm.phy.cam.ac.uk/
- Department of Materials, Imperial College London: https://www.imperial.ac.uk/engineering/departments/materials/
- Department of Chemistry, Southampton: http://www.southampton.ac.uk/chemistry/index.page
- Department of Physics, Warwick: http://www2.warwick.ac.uk/fac/sci/physics/
Publications
Publications describing the ONETEP code
J.C.A. Prentice, J. Aarons, J.C. Womack, A.E.A. Allen, L. Andrinopoulos, L. Anton, R.A. Bell, A. Bhandari, G.A. Bramley, R.J. Charlton, R.J. Clements, D.J. Cole, G. Constantinescu, F. Corsetti, S.M.-M. Dubois, K.K.B. Duff, J.M. Escartín, A. Greco, Q. Hill, L.P. Lee, E. Linscott, D.D. O’Regan, M.J. S. Phipps, L.E. Ratcliff, A.R. Serrano, E.W. Tait, G. Teobaldi, V. Vitale, N. Yeung, T.J. Zuehlsdorff, J. Dziedzic, P.D. Haynes, N.D.M. Hine, A.A. Mostofi, M.C. Payne and C.-K. Skylaris The ONETEP linear-scaling density functional theory program (2020) J. Chem. Phys. 152, 174111
J. Fox, J. Dziedzic, T. Fox, C. S. Tautermann and C.-K. Skylaris (2014). Density Functional Theory Calculations on Entire Proteins for Free Energies of Binding: Application to a Model Polar Binding Site. PROTEINS, 82, 3335.
Dziedzic, Q.O. Hill and C.-K. Skylaris (2013). Linear-scaling Calculation of Hartree-Fock Exchange Energy with Non-orthogonal Generalised Wannier Functions. Journal of Chemical Physics 139: 214103.
N.R.C. Corsini, N.D.M. Hine, P.D. Haynes, C. Molteni (2017) Unravelling the roles of size, ligands, and pressure in the piezochromic properties of CdS nanocrystals. Nano letters 17, 1042-1048
N.R.C. Corsini et al. (2015). Pressure-induced Amorphization and a New High Density Amorphous Metallic Phase in Matrix-free Ge Nanoparticles. Nano letters 15.11: 7334-7340.
Image 1: ONETEP simulation showing a defect in 1000 atoms of silicon. (Simulation produced by Dr Fabiano Corsetti)
Image 2:
Representations of the electron density (left) and kinetic energy density (right) for methane (centre), calculated by ONETEP – both of these quantities can be used to model electron interactions in DFT. (Simulation produced by Dr James Womack)
Image 3:
Excitations in the Fenna-Matthews-Olson complex – the zoom-in shows the electron-hole difference density of an excitation associated with Bacteriochlorophyll pigments, as computed by a ONETEP calculation on a 2000 atom fragment. (Simulation produced by Dr Tim Zuehlsdorff)
Header:
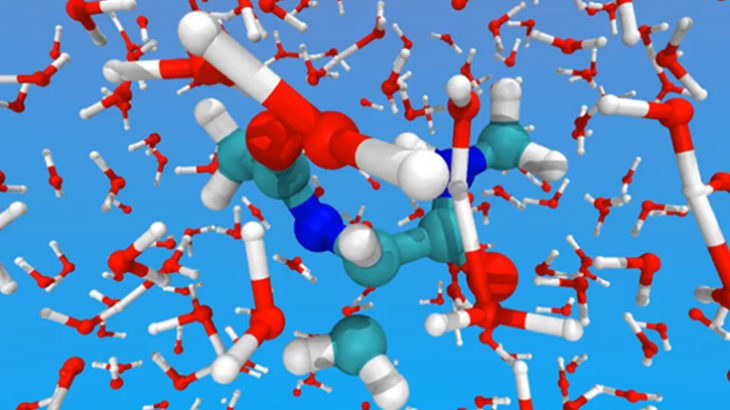
Snapshot from ab initio molecular dynamics (AIMD) simulation of peptide in water with ONETEP. (Simulation produced by Professor Chris-Kriton Skylaris)
Contacts
Professor Mike Payne
TCM Group, Cavendish Laboratory, University of Cambridge

Professor Chris-Kriton Skylaris
School of Chemistry, University of Southampton
Dr Jacek Dziedzic
School of Chemistry, University of Southampton
Dr Nicholas Hine
Department of Physics, University of Warwick
Professor Arash Mostofi
School of Chemistry, Imperial College London

Professor Peter Haynes
Department of Materials, Imperial College London